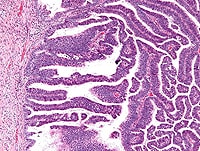
The major interest here at the CRBCM is of course obtaining a cure.
There is therefore a strong interest in knowing more about cancers that
are deemed curable today. Studying the genes known in these diseases
could provide some clues to their susceptibility to chemotherapy drugs
available today. Below are some of the genes for CHORIOCARCINOMA:
1. NECC1 on 4q11 (notice the "q" as this may be bad news)
This
is a gene of differentiation. And from what we have gathered, it is a
gene that codes for a Core Binding Factor like Molecule, these complexes
of major proteins with various functions put together to direct
cellular functions in directions. The proteins globally function as
regulators of other cellular functions. These proteins are sequentially
positioned in the CBF to drive into some directions. Most of the times,
the CBF has a portion that attaches to the DNA. The attachment could
silence the DNA. The silenced DNA here seems to block Cardiac
differentiation and forces the direction of activity toward syncytial
trophoblastic differentiation. This is why this gene is expressed in
normal placenta. In choricarcinoma, dedifferentiation occurs and NECC1
is mutated and silenced. This is called a suppressor gene as it relates
to Cardiac muscle differentiation, but clearly not for the tumor from
what we gathered. The Mutation will have to occur at the "q" location.
We will look further to establish if the "p" of this gene is located on
chromosome 7.
Desactivation of NECC1 leads to cardiac
hypertrophy. Being a CBF like molecule, it may solicit Histone deacetyl
transferases as part of its nuclear activity. We still are unclear
whether it is at the center of the pathogenesis of choriocarcinoma or
not!
------------------------------------------------------------------------------------------------------
2.CSH1
- The
protein encoded by this gene is a member of the somatotropin/prolactin
family of hormones and plays an important role in growth control. The
gene is located at the growth hormone locus on chromosome 17 along with
four other related genes in the same transcriptional orientation; an
arrangement which is thought to have evolved by a series of gene
duplications. Although the five genes share a remarkably high degree of
sequence identity, they are expressed selectively in different tissues.
Alternative splicing generates additional isoforms of each of the five
growth hormones, leading to further diversity and potential for
specialization. This particular family member is expressed mainly in the
placenta and utilizes multiple transcription initiation sites.
Expression of the identical mature proteins for chorionic
somatomammotropin hormones 1 and 2 is up-regulated during development,
although the ratio of 1 to 2 increases by term. Mutations in this gene
result in placental lactogen deficiency and Silver-Russell syndrome.
[provided by RefSeq, Jul 2008]"
===========================================================================
3. IGFR1
"Insulin-like growth factor 1 (
IGF-1), also called
somatomedin C, is a
protein that in humans is encoded by the
IGF1 gene.
[1][2] IGF-1 has also been referred to as a "
sulfation factor"
[3] and its effects were termed "nonsuppressible insulin-like activity" (NSILA) in the 1970s.
IGF-1 is a
hormone similar in
molecular structure to
insulin. It plays an important role in childhood growth and continues to have
anabolic effects in adults.
IGF-1 is produced throughout life. The highest rates of IGF-1
production occur during the pubertal growth spurt. The lowest levels
occur in infancy and old age.
Other IGFBPs are inhibitory. For example, both
IGFBP-2 and
IGFBP-5
bind IGF-1 at a higher affinity than it binds its receptor. Therefore,
increases in serum levels of these two IGFBPs result in a decrease in
IGF-1 activity."(Wikipedia)
IGFR1 amplification is used in autocrine faction to drive cancer growth, it acts here a as a TGF.
=======================================================================
4. CHFR
E3 ubiquitin-protein ligase CHFR is an
enzyme that in humans is encoded by the
CHFR gene.
[1][2][3]
(wikipedia)
"One protein that has been suggested to be part of the antephase
checkpoint is Chfr (checkpoint protein with an FHA domain and ring
finger;
Scolnick and Halazonetis, 2000), a ubiquitin ligase that is down-regulated in several cell lines through methylation of its promoter (
Mizuno et al., 2002). Chfr was originally reported to delay progress to prometaphase in the presence of colcemid (
Scolnick and Halazonetis, 2000), and cells were surprisingly described as delaying with high cyclin B1-Cdk1 activity (
Scolnick and Halazonetis, 2000),
which conflicted with a role as part of the antephase checkpoint
because cyclin B1-Cdk1 is fully activated only in late prophase.
However, in
Xenopus laevis extracts, Chfr is able to delay the
activation of cyclin B-Cdk1, apparently by targeting the Polo-like
kinase, Plx, for degradation by the proteasome (
Kang et al., 2002),
thereby preventing the activation of the Cdc25 phosphatase that
activates Cdk1. Chfr has also been reported to affect Polo-like kinase
levels in human cells in response to DNA damage (
Shtivelman, 2003).
But whether Chfr does target Polo for degradation or not is debatable
because Chfr has been shown to conjugate ubiquitin via its lysine 63
residue (
Bothos et al., 2003) that normally acts in signal transduction, especially for stress signals (
Deng et al., 2000;
Ulrich and Jentsch, 2000;
Hofmann and Pickart, 2001;
Pickart, 2001;
Wang et al., 2001), rather than to target proteins to the proteasome."
Matsusaka and Pines suggested.
CERTAINLY DOWN REGULATION HERE OPEN THE DOOR TO USE OF VELCADE OR THE PROTEASOME INHIBITORS.
--------------------------------------------------------------------------------------------------------
5. MUC3A"Associations of distinct variants of the intestinal mucin gene MUC3A with
ulcerative colitis and
Crohn's disease [1]." (WIKI) Its gene is located at 7q22 (notice the q) it has a prognosis value
But
we also know that MUC1 will be more important in this cancer and shield
by its mucinous product cell from cancer watching cells.
James
Gum et al "Mucinous cancers are generally more extensive at diagnosis.
Membrane mucins are an important class of glycoproteins with diverse
structures and functions. These molecules contain extracellular
domains that serve as a scaffold for
O-glycosylation. The
O-glycans
associated with membrane mucins are thought to function in
cytoprotection and have been demonstrated to confer anti-adhesion
properties upon cells (
1).
This latter characteristic may play a role in the dissemination and
spread of cancer cells. In addition to conferring these
electrostatic/physical properties upon cells,
membrane mucins can anchor carbohydrate moieties with specific
functions. Selectin
ligands associated with membrane mucin glycans, for
example, play a role in cancer cell extravasation during metastases (
2). Certain membrane mucins function in signal transduction as well (
3–
5). Several membrane mucins also serve as clinically important tumor antigens (
6,
7)."
--------------------------------------------------------------------------------------------------------------
6. TAF7
TAF7
From Wikipedia, the free encyclopedia
Transcription initiation factor TFIID subunit 7 also known as TAFII55 is a
protein that in humans is encoded by the
TAF7 gene.
[1]
The intronless gene for this transcription coactivator is located
between the protocadherin beta and gamma gene clusters on chromosome 5.
The protein encoded by this gene is a component of the TFIID protein
complex, a complex which binds to the TATA box in class II promoters and
recruits RNA polymerase II and other factors. This particular subunit
interacts with the largest TFIID subunit, as well as multiple
transcription activators. The protein is required for transcription by
promoters targeted by RNA polymerase II.
[2]
The general
transcription factor, TFIID, consists of the TATA-binding
protein (TBP) associated with a series of TBP-associated factors (TAFs) that together participate in the assembly of the
transcription preinitiation
complex. TAFII55
binds to TAFII250 and
inhibits
its acetyltransferase activity. The exact role of TAFII55 is currently
unknown but studies have shown that it interacts with the
C-jun pathway.
[3] The
conserved region is situated towards the N-terminal of the protein.
[4] This entry talks about the
N-terminal domain.
TAF7 interacts with TATA which
The
TATA-binding protein (
TBP) is a
general transcription factor that binds specifically to a
DNA sequence called the
TATA box. This DNA sequence is found about 30
base pairs upstream of the
transcription start site in some
eukaryotic gene promoters.
[1] TBP, along with a variety of
TBP-associated factors, make up the
TFIID, a
general transcription factor that in turn makes up part of the
RNA polymerase II preinitiation complex.
[2]
As one of the few proteins in the preinitiation complex that binds DNA
in a sequence-specific manner, it helps position RNA polymerase II over
the
transcription start site
of the gene. However, it is estimated that only 10-20% of human
promoters have TATA boxes. Therefore, TBP is probably not the only
protein involved in positioning RNA polymerase II.
TBP is involved in
DNA melting (double strand separation) by bending the
DNA
by 80° (the AT-rich sequence to which it binds facilitates easy
melting). The TBP is an unusual protein in that it binds the minor
groove using a β sheet.
Another distinctive feature of TBP is a long string of glutamines in
the N-terminus of the protein. This region modulates the DNA binding
activity of the C-terminus, and modulation of DNA-binding affects the
rate of transcription complex formation and initiation of transcription.
Mutations that expand the number of CAG repeats encoding this
polyglutamine tract, and thus increase the length of the polyglutamine string, are associated with
spinocerebellar ataxia 17, a
neurodegenerative disorder classified as a
polyglutamine disease.
[3]
BASICALLY INITIATE OR PARTICIPATE IN THE INITIATION OF OF TRANSCRIPTION/TRANSLATION
--------------------------------------------------------------------------------------------------------------
8. CDC123
9. PSMD
10. HAS2
11. CD44 a member of the EZRIN/S100P/ Villin -Complex
------------------------------------------------------------------------------------------------------
12. S100P
S100P
From Wikipedia, the free encyclopedia
Protein S100-P is a
protein that in humans is encoded by the
S100P gene.
[1][2][3]
The protein encoded by this gene is a member of the
S100 family of proteins
containing 2 EF-hand calcium-binding motifs. S100 proteins are
localized in the cytoplasm and/or nucleus of a wide range of cells, and
involved in the regulation of a number of cellular processes such as
cell cycle progression and differentiation. S100 genes include at least
13 members which are located as a cluster on chromosome 1q21; however,
this gene is located at 4p16. This protein, in addition to binding Ca
2+, also binds Zn
2+ and Mg
2+. This protein may play a role in the etiology of prostate cancer.
[3]
Interactions
S100P has been shown to
interact with
EZR.
[4] which this protein serves as an intermediate between the plasma membrane and the
actin cytoskeleton. It plays a key role in cell surface structure adhesion, migration, and organization.
[2]
Interactions
VIL2 has been shown to
interact with
Sodium-hydrogen exchange regulatory cofactor 2,
[3][4] Merlin,
[5] SDC2,
[6] CD43,
[7] Fas ligand,
[8][9] VCAM-1,
[10] S100P,
[11] ICAM3,
[12][13] ICAM-1,
[12] Sodium-hydrogen antiporter 3 regulator 1,
[14][15] ICAM2,
[12] Moesin,
[8][16][17] PALLD[18] and
PIK3R1.
[19]
Given
the multitude of interactions the EZERIN appears a critical molecules
for signal propagation intra and extra-cellularly. EZRIN the Villin
(Vilain) comes into everything. In the endothelium, This complex of molecules is " involved in the generation and maintenance of the anchoring
structure. These results provide the first characterization of an
endothelial docking structure that plays a key role in the firm adhesion
of leukocytes to the endothelium during inflammation."
This is a basis for novel anti-inflammatory strategy.
It
also interact with the so called Peripheral proteins of which "some
are water-soluble proteins and associate with lipid bilayers
irreversibly and can form transmembrane alpha-helical or
beta-barrel channels. Such transformations occur in
pore forming toxins such as
colicin A, alpha-hemolysin, and others. They may also occur in
BcL-2 like protein , in some amphiphilic
antimicrobial peptides , and in certain
annexins
. These proteins are usually described as peripheral as one of their
conformational states is water-soluble or only loosely associated with a
membrane.
[11]"
----------------------------------------------------------------------------------------------------
13. delta-like1
14. STOX1
15. JAK/STAT1
16. GCM1
Chorion-specific transcription factor GCMa is a
protein that in humans is encoded by the
GCM1 gene.
[1][2]
This gene encodes a DNA-binding protein with a gcm-motif (glial cell
missing motif). The encoded protein is a homolog of the Drosophila glial
cells missing gene (gcm). This protein binds to the GCM-motif
(A/G)CCCGCAT, a novel sequence among known targets of DNA-binding
proteins. The N-terminal DNA-binding domain confers the unique
DNA-binding activity of this protein.
[2]WIKIPEDIA
Chou et al "the activity of GCMa can be post-translationally regulated by protein
phosphorylation,
ubiquitination, and acetylation, it is unknown
whether GCMa activity can be regulated by sumoylation. In this report,
we investigated
the role of sumoylation in the regulation of GCMa
activity. We demonstrated that Ubc9, the E2 component of the sumoylation
machinery...Our study
demonstrates that GCMa is a new sumoylation
substrate and its activity is down-regulated by sumoylation."
-----------------------------------------------------------------------------------------------------------
18.
Cx31: Connexin, important in cell adhesion and embryogenesis. In adult
Mutation at Cx31 lead to sensory neural deafness as it contribute to
synaptic integrity and Monocyte. A legitimate secondary target in blood
disease.
-----------------------------------------------------------------------------------------
also we will include
19. Endoglin
20. Syncitin
21. HCG
22. Connexin 31
23. E-Cadherin
Lets go to work!
Another thing to look at is the propensity in these diseases to develop thrombosis on BEP treatment!
---------------------------------------------------------------------------------------------------
Basically we see an emphasis on
1. Trophoblastic differentiation
2. a drive to dupplication and genetic amplification
3. Use of IGF as autocrine growth hormone
4. use of invasive and adhesion molecule to metastasize a prominent feature of Choriocarcinoma
5. NF2 could be the indicator of prominent tendency to brain metastasis, we will look for it in Breast and small cell lung cancer
6. E2 Methylation introduces Velcade an the anti-proteasome in refractory disease
7 Anti-EZRIN/Villin as a strategy
8. somewhere, where is the WNT pathways in all this?
Chemotherapy cures here because of rapid DNA multiplication!